ACS Catalysis│甲苯单加氧酶控制底物选择性的分子基础
今天推送的文章发表在ACS Catalysis的“Molecular Basis for a Toluene Monooxygenase to Govern Substrate Selectivity”,通讯作者为湖北大学生命科学学院,生物催化与酶国家重点实验室的李爱涛教授。
细胞色素 P450 单加氧酶(P450s 或 CYPs)属于血红素氧化酶的超家族,以区域选择性和立体选择性的方式催化非活性 C-H 键的氧化反应,从而实现化学方法通常难以实现的化学转化。苯甲醇 (2a) 是是一种重要的化学品,一般由甲苯 (1a)羟基化生成,通常用作合成药物、香料和杀虫剂的中间体。目前苯甲醇的主要经过能源密集且危险的两步化学反应生产,导致环境问题(方案1A)。
应用P450可以克服这一问题,一些 P450单加氧酶表现出甲苯羟基化活性,但其中大多数的选择性或/和活性不高。自给自足的P450LaMO仅以24.4%的转化率将1a转化为2a,同时形成副产物苯甲醛。P450BM3催化甲苯苯环邻位羟基化,生成99%的邻甲酚,不生成苯甲醇。P450BM3的突变体KT5 (F87A/A330P/E377A/D425N) 则主要生成苯甲醇2a,但仍产生5%的邻甲酚。来自Novosphingobium的P450CYP101B1及其突变H85F催化甲苯仅产生苯甲醇,但活性极低(转化率 2-4%)。

在之前的研究中,作者从甲苯降解菌株Rhodococcus coprophilusTC-2中鉴定出一种P450(P450tol),能够羟基化甲苯且只生产苯甲醇(方案1B),是迄今为止已知的唯一一种天然衍生的甲苯羟基化酶,但缺乏结构信息以及未确定其天然氧化还原伴侣,其催化分子机制仍未被阐明。在本研究中,作者报道了P450tol与底物甲苯1a和产物苯甲醇2a复合物的晶体结构,以阐明底物结合模式和催化机理。为了促进其在生物转化中的应用,将P450tol的血红素结构域与来自红球菌属的P450RhF或来自嗜热Tepidiphilus thermophiles的CYP116B46还原酶结构域融合,以产生电子传递链自给自足的酶。最后对热稳定性和活性改善的突变体进行表征并评估了其转化甲苯1a、卤代甲苯 (1b-1f)以及丙苯 (3)的能力。
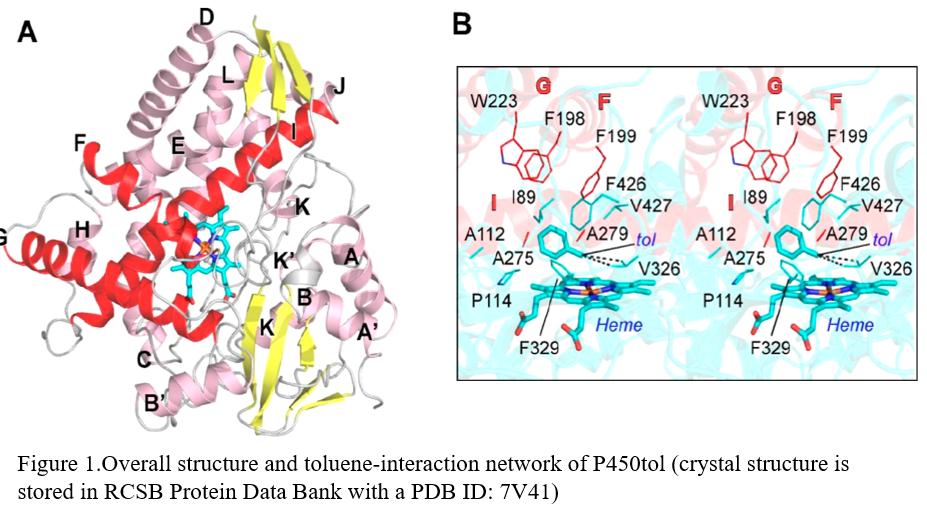
对P450tol的结构分析和阐明配体结合模式
在三角晶胞中结晶P450tol重组蛋白,与其他经典P450 类似,P450tol的整体结构主要包含螺旋,血红素辅基硫醇与严格保守的C末端C389残基连接(图1A)。P450tol与结构最接近的P450terp的序列同一性为46%,其结构最明显的变化在于血红素远端上方的螺旋G和F(图S1),这两个螺旋控制底物进入隧道。先前报道的P450terp结构显示开放构象,而P450tol结构似乎显示出封闭构象(图S1)。

P450tol-甲苯复合物和apo形式的结构几乎相同 (Cα RMSD, 0.05 Å)。配体位于血红素远端的疏水口袋中,被 I89、A112、P114、F198、F199、W223、A275、A279、F329、F426 和V427包围。甲苯羟基化位点的甲基朝向血红素铁(图1B),F329和A279通过T形堆叠效应夹住甲苯苯环,V326的侧链位于甲基前∼4 Å(图1B),这些氨基酸有助于将底物保持在观察位置,使羟基化可以精确地发生在苯甲基位点上。P450tol-苯甲醇2a复合物的结构中有相似的结合位置和相互作用。包括F199、F426、A279、V326和F329在内的几个底物相互作用残基在P450tol同源序列中严格保守,表明它们在其他P450tol相关酶中可能起关键作用。

对甲苯结合残基的突变研究
结构分析表明,至少有三个氨基酸(F329、A112和V326)控制P450tol催化甲苯羟基化的区域选择性。作者进行了诱变实验以验证这三个甲苯结合残基的作用。首先,研究了严格保守的残基F329在与甲苯苯环T形堆积相互作用中的功能,对F329进行了饱和诱变并获得了19个突变体。活性检测显示,大多数突变体失去了对甲苯的活性,只有突变体F329M保留了42%的活性(图2A),表明Phe在该位置至关重要。位于活性位点口袋的几个残基,包括A112、V326和F427也被靶向并突变为其他不同大小的疏水残基以进行活性测试。如图2B 所示,所有突变体的活性都大大降低,V326G和V427G完全没有活性,这证明关键氨基酸对于维持P450tol酶活性非常重要。

构建人工嵌合酶
与I类P450单加氧酶相比,血红素结构域与氧化还原伴侣自然融合的电子传递链自给自足P450单加氧酶由于有效的电子转移效率而表现出较高的活性,例如P450BM3和P450RhF。P450RhF的还原酶结构域 (RhFRED)已被证明是一种有效的氧化还原伴侣,当与I类P450融合时可产生具有改进催化活性的嵌合酶。受此启发,作者尝试将还原酶RhFRED与P450tol的血红素结构域融合以产生自给自足的P450嵌合体。

P450tol和还原酶 (FMN/FeS) 结构域之间的接头区域在电子转移中起重要作用,影响嵌合酶的催化活性。以天然接头连接的P450tol-RhFRED作为对照,作者构建了九种含有不同长度的人工接头的嵌合酶(
P450tol-RhFRED-L1-L7-P1-P2,图3)。经过表达后,含有相应嵌合酶的全细胞裂解物被用于甲苯羟基化反应。除P450tol-RhFRED L2之外的所有嵌合酶都表现出比P450tol-RhFRED更高的活性,其中P450tol-RhFRED L3的活性最高,提高了4.7倍以上(图4)。为增强蛋白质稳定性,又选择从嗜热嗜热菌中新发现的嗜热自给自足P450CYP116B46的还原酶结构域构建嵌合酶。在25 °C下,P450tol-CYP116B46在20h时保持90%以上的活性(图5)。

相比之下,P450tol-RhRED L3在6小时后失去活性。P450tol-CYP116B46的活性半衰期比P450tol-RhRED L3更长(68.5h对2.7h)并且Tm增加了14°C(表1)。由于具有相同的底物结合区域,这两种酶显示出相似的底物结合亲和力(表1)。与P450tol-RhFRED L3相比,P450tol-CYP116B46的催化性能 (kcat/Km)提高了两倍以上(表 1)。因此,利用结构域置换策略成功获得了嗜热融合酶P450tol-CYP116B46。

P450tol对卤代甲苯的催化活性
由于从甲苯复合结构中观察到与V326定位侧相对的额外空间(图1B),因此P450tol可能催化较大甲苯衍生物的羟基化。作者使用两种P450tol嵌合酶测试了卤代甲苯(1b-1f)是否可以被P450tol区域选择性羟基化。结果表明,尽管TTN低于甲苯1a, 1b-1f都可以在甲基上羟基化(表2),P450tol-CYP116B46表现出比P450tol-RhRED L3高1.8-5.1倍的活性。对于对位取代底物,两种嵌合酶对氟甲苯的活性最高 (F > Br > Cl,表2)。对于氯代甲苯,两种嵌合酶对氯甲苯1d的活性最高,其次是间氯甲苯和邻氯甲苯(表2)。

随后作者用各种卤代甲苯化合物浸泡P450tol晶体,获得了P450tol与对氯甲苯1d、间氯甲苯1c、邻氯甲苯1b和对溴甲苯1e的复合物(图S14)。除邻氯甲苯1b外,所有化合物都与P450tol结合,其苯环位于F329和A279之间,侧甲基朝向血红素铁,与甲苯配合物相同,这些化合物的结合模式显示出有利于生产反应的构象。而邻氯甲苯1b有不同的结合模式,甲基指向与V326相对的方向,苯环面向血红素-Fe。但由于邻氯甲苯1b也可被P450tol在苯甲基位点羟基化,观察到的结合姿势可能不能反映生产反应中的状态。

工程化P450tol-CYP116B46用于丙苯的区域和立体选择性羟基化
最后,作者探讨了P450tol-CYP116B46区域选择性和立体选择性羟基化具有脂肪族侧链的芳香族化合物丙苯中的潜力。如图6A所示,四种对映体纯羟基化产物 (S)-或 (R)-4;(S)-或(R)-5原则上可以通过亚末端或苯甲基羟基化生成,这些醇是药物合成中非常有用的中间体。在先前研究中,P450pyr被设计用于催化丙苯,能够提供具有95% ee和98%亚末端选择性的 (S)-4;P450BM3 WT以90%的ee和99%的苯甲基选择性产生 (R)-5,而P450BM3突变体KT5以78%的选择性产生1-苯基-2-丙醇 (4)。

在本研究中,首先使用野生型P450tol-CYP116B46催化3羟基化,主要产物为 (S)-4,并显示出严格的亚末端选择性,但ee只有37%。为了进一步提高立体选择性并将亚末端羟基化转变为苯甲基羟基化以生产1-苯基-1-苯基丙醇(5),基于P450tol的结构-功能关系作者进行了进一步的蛋白质工程。通过将3停靠在P450tol的结合口袋中确定了3周围的关键残基,包括F329、P114和V326(图6B)。由于F329对于维持P450tol的羟基化活性至关重要,因此只对位于底物前后的P114和V326进行迭代饱和诱变 (ISM) 。对P114饱和诱变文库筛选后,突变体P114A对(S)-4的立体选择性从37.0提高到86.3%(图6C和D)。另外,V326突变体库中的V326C区域选择性发生改变,羟基化主要发生在苯甲基位置,以89% ee生成 (S)-5(苯甲基区域选择性为82.3%,图6C和D)。为了进一步提高V326C的区域选择性和立体选择性,对P114进行了第二轮饱和诱变。经过筛选,获得了双变体V326C/P114V,其 (S)-5的ee率为96.4%,苯甲基区域选择性为94.2%。改变的区域选择性可能是由于第326位残基的较小侧链产生更大的空间来容纳丙苯的烃部分,从而导致羟基化位置从亚末端转移到苯甲基位点。
整理:梁子琦
DOI::10.1021/acscatal.1c05845
